Ученые МФТИ определили ключевые гены-драйверы онкологии
В настоящее время науке известны онкогенные мутации в более чем 300 генах, и зачастую возникает необходимость ранжировать их, чтобы определить мишени для основного воздействия при лечении. Но в большинстве случаев мутации ранжируются по частоте встречаемости, что необязательно соответствует их значимости в заболевании. Взять за основу силу мутаций предложили ученые МФТИ.

С помощью биоинформатического анализа крупнейшей базы данных об онкогенных мутациях TCGA PanCanAtlas они разработали новый способ ранжировать гены-драйверы, запускающие болезнь. Результаты исследования опубликованы в научном журнале PeerJ Life & Environment.
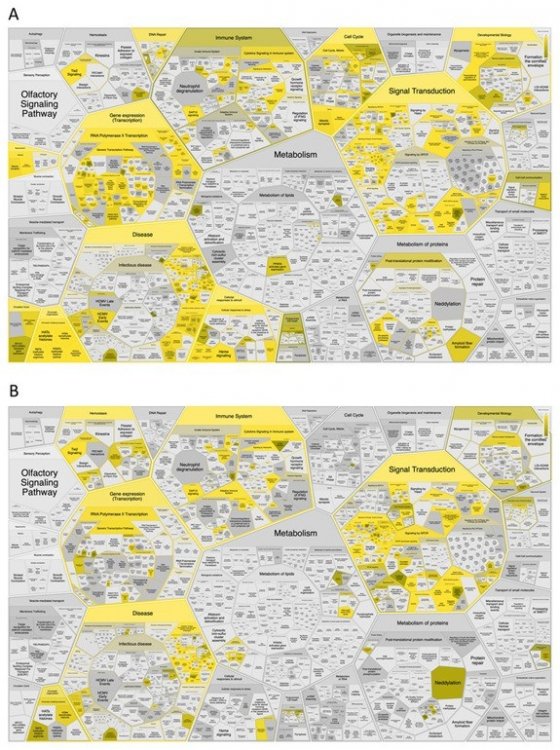
Статистически значимое представительство в функциональных категориях 50 наиболее сильных драйверных генов по версии Индекса силы драйвера (А) и Нормализованного индекса силы драйвера (В) — желтый цвет указывает на статистическую значимость.
Согласно господствующей сегодня точке зрения, рак инициируется и стимулируется мутациями в так называемых «драйверных» генах, в то время как мутации в «пассажирских» генах не оказывают никакого влияния и просто «путешествуют» вместе с драйверами в ходе соматической эволюции. Существует несколько подходов к выявлению мутаций-инициаторов рака и их ранжированию, основные из них — по частоте встречаемости, по эффекту на трехмерную структуру белка и по эффекту на взаимодействие белков. У этих способов есть свои плюсы и минусы.
Один из подходов заключается в использовании частоты мутаций среди пациентов с определенным видом рака, обычно с поправкой на частоту фоновых мутаций в гене. Этот способ действительно позволяет выявить наиболее распространенные мутации, но это не означает, что они способны самостоятельно вызвать рак. В то же время мутация может быть редкой, но достаточной для инициации рака. Первый случай будет примером распространенного, но слабого драйвера, тогда как второй будет редким, но сильным драйвером. Таким образом, алгоритмы, основанные на частоте встречаемости мутаций, не могут надежно определять силу генов-драйверов.
Существует большая группа алгоритмов, которые нацелены на прогнозирование и ранжирование генов-драйверов в соответствии с влиянием мутаций на структуру и активность белков. Эти методы могут определить, нарушены ли структура и функция белка и в какой степени, однако они гораздо меньше подходят для определения роли конкретного белка в контексте других белков клетки и ее микроокружения. А именно это имеет решающее значение для определения того, будет ли он вызывать рак. Таким образом, степень влияния мутации на структуру белка не определяет того, является ли этот белок онкогенным.
Третий подход определяет влияние мутаций на взаимодействие белков, выявляя ключевые молекулы с наибольшим количеством взаимодействий с другими белками в клетке. Подразумевается, что мутации в таких белках будут иметь самые серьезные последствия для клетки. Обычно так и оказывается, но гораздо чаще это приводит к гибели клетки, чем к онкогенной трансформации. Таким образом, данный способ также не подходит для ранжирования мутаций по их силе.
«Ранее мы провели количественную оценку драйверных мутаций и обнаружили очень высокую вариабельность в числе мутаций даже среди пациентов с одним и тем же типом рака. Поэтому мы задались вопросом: в чем причина того, что у некоторых пациентов достаточно одной драйверной мутации, тогда как у других рак не развивается до тех пор, пока не накопятся десятки мутаций? Мы предположили, что основной причиной является их сила: один сильный драйвер может быть эквивалентен по своей опухоль-индуцирующей активности нескольким слабым.
Следовательно, статистически более вероятно, что сильные драйверы встречаются чаще у пациентов с малым количеством мутаций, ведь нескольких сильных драйверных мутаций достаточно, чтобы инициировать рак. И наоборот, слабые драйверы чаще встречаются у пациентов с большим числом драйверных мутаций, потому что слабых драйверов для инициации рака нужно много. На основе этого принципа мы разработали математические формулы, которые позволили нам создать биоинформатические алгоритмы для автоматического вычисления численного эквивалента силы любой драйверной мутации на основании ее распределения среди пациентов с разным числом драйверных мутаций», — рассказал ведущий автор исследования Алексей Беликов.
Источник: https://scientificrussia.ru/
9.09.2022